


 雷达卡
雷达卡



为何要进行降维处理?
在单细胞转录组研究中,每个细胞通常包含数千个基因的表达信息,导致数据维度极高。为了更高效地处理和分析这些高维数据,降维成为关键步骤。其主要目标是将原始高维空间映射到一个低维空间,从而实现以下目的:
- 降低噪声干扰:通过压缩数据维度,剔除技术性噪音,保留具有生物学意义的信息。
- 便于数据可视化:将高维数据投影至二维或三维空间,有助于直观识别细胞亚群之间的分布模式与差异。
- 提升聚类与分类效果:在低维空间中,细胞间的相似性更容易被捕捉,有利于后续的聚类分析和细胞类型鉴定。
- 提高计算效率:减少数据维度后,可显著降低后续分析(如聚类、轨迹推断等)所需的计算资源与时间开销。
常见的三种降维方法对比
| 方法 | 类型 | 适用场景 | 优点 | 缺点 |
|---|---|---|---|---|
| PCA | 线性降维 | 初步降维、数据预处理 | 计算速度快,解释性强,适用于大规模数据集 | 仅能捕捉线性结构,对非线性关系建模能力弱 |
| t-SNE | 非线性降维 | 数据可视化、局部结构展示 | 擅长揭示复杂的非线性结构,广泛用于细胞群体的可视化呈现 | 运算耗时较长,不适用于超大数据集;结果具有随机性,重复性较差 |
| UMAP | 非线性降维 | 可视化及保持全局与局部结构 | 运行效率高于t-SNE,稳定性更好,能同时保留数据的全局与局部特征,适合大样本分析 | 对参数设置敏感,需进行调参优化以获得理想结果 |
实际操作流程
# PCA 是基于 HVG
seob <- RunPCA(seob, assay =?"SCT")
# s-SNE 不是基于表达矩阵做的,而是基于PCA结果
?seob <- RunTSNE(seob, assay =?"SCT", dims = 1:30)?
# UMAP
seob <- RunUMAP(seob, assay =?"SCT", dims = 1:30)
#可视化
p_pca <- DimPlot(seob,?
? ? ? ? reduction =?"pca",?# pca, umap, tsne
? ? ? ? group.by =?"samples",
? ? ? ? label = F)
p_umap <- DimPlot(seob,?
? ? ? ? reduction =?"umap",?# pca, umap, tsne
? ? ? ? group.by =?"samples",
? ? ? ? label = F)
p_pca + p_umap数据整合与批次效应去除
当多个实验批次的数据存在系统性偏差时,即出现“批次效应”,可能掩盖真实的生物学差异。因此,在合并不同来源的数据前,需要进行整合处理以消除此类影响。
常用整合方法包括:
- CCAIntegration
- RPCAIntegration
- HarmonyIntegration
- FastMNNIntegration
- scVIIntegration
选择依据如下:
- 数据规模:对于较大规模数据集,推荐使用 RPCAIntegration、HarmonyIntegration 或 FastMNNIntegration;面对超大规模数据,scVIIntegration 表现尤为出色。
- 模态类型:若涉及多组学数据(如 RNA 和 ATAC-seq),CCAIntegration 与 scVIIntegration 更具优势。
- 批次效应强度:HarmonyIntegration 与 FastMNNIntegration 在缓解批次效应方面表现优异。
- 计算资源限制:若硬件资源有限,RPCAIntegration 是高效的轻量级选项;而 scVIIntegration 可充分利用 GPU 加速,适合具备高性能计算环境的用户。
注意:FastMNNIntegration 不支持 SCTransform 标准化流程,需采用传统标准化方式。此外,scVIIntegration 强烈建议配备 GPU 支持以提升训练速度。
参考文档:Seurat 官方整合指南
实操示例
seob_harmony <- IntegrateLayers(object = seob,?
? ? ? ? ? ? ? ? ? ? ? ? normalization.method =?"SCT",
? ? ? ? ? ? ? ? ? ? ? ? orig.reduction =?"pca",?
? ? ? ? ? ? ? ? ? ? ? ? method = HarmonyIntegration,?
? ? ? ? ? ? ? ? ? ? ? ? new.reduction =?"integrated.harmony")
#重新降维
seob_harmony <- RunUMAP(seob_harmony, assay =?"SCT", dims = 1:30,
? ? ? ? ? ? ? ? reduction =?"integrated.harmony")?
DimPlot(seob_harmony,?
? ? ? ? reduction =?"umap",?
? ? ? ? group.by =?"samples",
? ? ? ? label = F)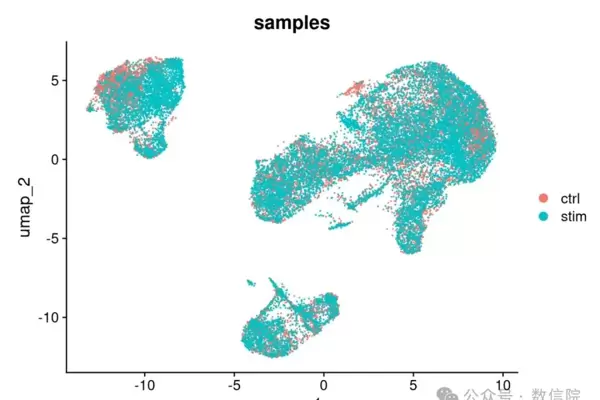
提示:是否执行批次整合应根据具体研究设计判断。不当整合可能导致真实生物学差异被误校正而丢失,尤其在比较不同处理组或疾病状态时需谨慎操作。
聚类分析流程详解
聚类的第一步是构建细胞间的关系图谱。Seurat 中的 FindNeighbors 函数用于计算每个细胞的 k 个最近邻,并可进一步生成共享近邻图(SNN)。该过程基于 Jaccard 指数衡量两个细胞邻域的重叠程度,进而反映其相似性,为后续聚类提供基础网络结构。
关键参数说明:
- k.param:定义最近邻数量,默认值为 20,即为每个细胞寻找最接近的 20 个邻居。
- nn.method:指定搜索算法,可选 rann 或 annoy。其中 annoy 属于近似最近邻算法,更适合处理大规模数据集。
- dims:指定参与构建近邻图的主成分维度,通常选用 PCA 结果中的前若干个主成分。
seob <- FindNeighbors(seob, reduction =?"pca", dims = 1:20)接下来,FindClusters 函数利用构建好的 SNN 图,采用模块性优化算法(如 Louvain 或 Leiden)来划分细胞簇。该算法通过最大化模块性得分,将图中连接紧密的节点划分为同一簇。
核心参数设置:
- resolution:控制聚类分辨率。数值越高,生成的细胞簇越多且更细粒度。
- algorithm:可选 Louvain(默认)或 Leiden。后者在分割精度和收敛性上更优,尤其适用于大数据集,但需预先安装 Python 包
leidenalg。
seob <- FindClusters(seob,
? ? ? ? ? ? ? ? ? ? ?resolution = 0.4,?# 值越大,cluster 越多
? ? ? ? ? ? ? ? ? ? ?random.seed = 1)?
DimPlot(seob,?
? ? ? ? reduction =?"umap",?
? ? ? ? group.by ?= c("seurat_clusters"),
? ? ? ? label = T)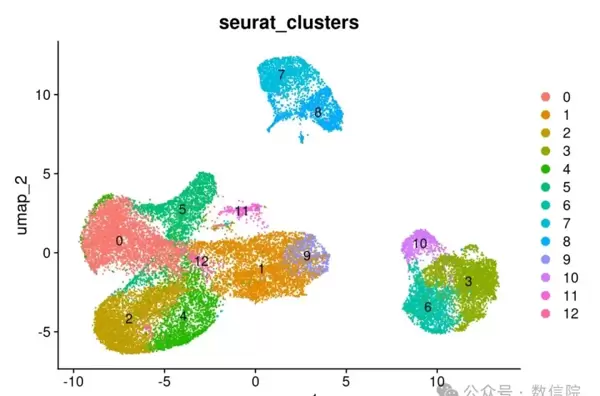
5. 采用自主采购模式,全部硬件为自购的裸金属服务器,部署在华东地区排名前50的IDC机房,接入省级骨干网络专线。

 京公网安备 11010802022788号
京公网安备 11010802022788号







