


 雷达卡
雷达卡



1. 富集分析基础概述
1.1 富集分析的定义与意义
富集分析(Enrichment Analysis)是生物信息学中的核心分析手段之一,主要用于判断在特定分子集合(如差异表达基因、蛋白质或代谢物)中,某些功能类别或生物学通路是否出现显著聚集现象。该方法广泛应用于转录组、蛋白质组及代谢组等高通量数据分析中,有助于揭示潜在的生物学机制、疾病相关通路以及基因功能注释。
其主要目标在于评估一组选定的生物分子(例如实验中筛选出的差异基因)是否在某个已知的功能集合中非随机地集中出现。举例来说,在研究某种癌症的基因表达谱时,若发现大量差异基因集中于某一信号通路,则通过富集分析可识别该通路是否具有统计学上的显著性,从而提示其可能参与疾病的调控过程。
1.2 常见的富集分析类型
目前主流的富集分析方法包括以下几类:
- GO富集分析:基于Gene Ontology数据库,从三个维度系统描述基因功能:
- 分子功能(Molecular Function, MF)——指基因产物在分子层面执行的具体活性,如酶催化或结合能力;
- 细胞组分(Cellular Component, CC)——表示基因产物在细胞内的定位区域,如线粒体、细胞核等;
- 生物过程(Biological Process, BP)——反映基因所参与的完整生物学活动,如细胞周期、免疫应答等。
- KEGG富集分析:依托京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes)数据库,对目标基因集进行通路注释和统计检验,识别其显著关联的代谢或信号传导通路,帮助理解基因在系统层面的功能角色。
- GSEA富集分析
- GSVA富集分析
本部分内容将重点介绍GO与KEGG两类富集分析的基本原理及其实际应用。GSEA和GSVA等相关方法将在后续章节中进一步展开说明。
1.3 如何解读富集分析结果
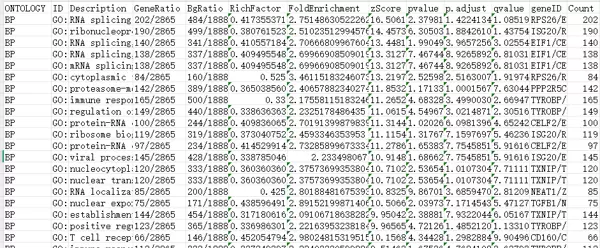
(1)GO富集分析结果解析
在GO富集结果表格中,各字段含义如下:
- ONTOLOGY:标明该条目所属的GO分类,即BP(生物过程)、MF(分子功能)或CC(细胞组分)。
- ID:GO通路的唯一编号,用于在数据库中精确检索对应条目,类似于“身份证号”。
- Description:对该GO条目的简要功能描述,便于快速理解其生物学意义。
- GeneRatio:表示富集到该通路的基因数与输入总基因数的比例,体现该通路在目标基因集中的覆盖程度。
- BgRatio:背景基因集中与该通路相关的基因占比,用于对比目标集与全基因组之间的分布差异。
- Pvalue:原始p值,衡量富集显著性的指标,数值越小表示越显著,通常以<0.05为阈值判断显著性。
- p.adjust:经过多重假设检验校正后的p值(常用Benjamini-Hochberg法),控制假阳性率,提供更严谨的显著性评估。
- Qvalue:即q值,用于估计假发现率(FDR),值越低说明富集结果越可靠。
- geneID:列出所有富集至该通路的基因ID,以斜杠分隔,可用于后续验证或网络构建。
- Count:实际映射到该通路上的基因数量,反映该通路被激活或影响的程度。
(2)KEGG富集分析结果解读
KEGG分析结果通常包含以下列信息:
- Category:通路所属的大类划分,如“Metabolism”(代谢)、“Genetic Information Processing”(遗传信息处理)或“Human Diseases”(人类疾病),便于宏观把握功能方向。
- Subcategory:子分类层级,进一步细化通路归属,例如在“Human Diseases”下可细分为“Neurodegenerative disease”(神经退行性疾病)等。
- ID:KEGG通路的唯一标识符,可在KEGG官网中直接查询详细通路图谱。
- Description:对通路功能的文字描述,帮助用户快速了解其生物学背景。
- GeneRatio:目标基因集中富集于该通路的基因比例,体现该通路在研究集合中的相对重要性。
- BgRatio:在整个参考基因组中,属于该通路的基因所占比例,作为比较基准。
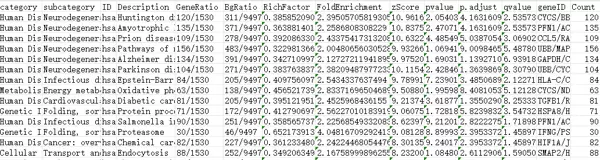
Pvalue:表示常规的 p 值,用于评估基因富集结果的统计显著性。p 值越低,说明该通路或功能类别的富集越显著。一般认为当 p 值小于 0.05 时,富集具有统计学意义。
p.adjust:为经过多重假设检验校正后的 p 值,常采用 Benjamini-Hochberg 方法进行计算,目的是控制整体的假阳性率。相比原始 p 值,校正后的 p 值在判断显著性时更为严格,能更可靠地反映真实富集情况。
Qvalue:即 q 值,是基于假发现率(False Discovery Rate, FDR)校正后的一种显著性指标,本质上也反映了校正后的显著性水平。q 值越小,表明富集结果中出现假阳性的可能性越低,富集越可信。
geneID:列出的是参与该通路或功能类别富集分析的基因标识符(Gene ID),各基因间以斜杠分隔。这些基因可用于后续的功能验证、网络构建或其他生物信息学分析。
Count:指在当前通路中被富集到的基因数量,直观体现有多少差异表达基因参与到该生物学过程或信号通路中。
2. GO 与 KEGG 富集分析
2.1 GO 富集分析
本部分将基于《转录组基因表达差异分析全流程:以GSE65682为例》一文中获得的差异表达基因结果,开展 GO 功能富集分析。
library(clusterProfiler)
library(org.Hs.eg.db)
导入要富集分析的基因
# 读取基因数据文件,文件中包含基因符号(gene symbols)
gene_data <- read.csv("D:\\Users\\Desktop\\转录组\\DEGhc_Sepsis.csv") ?
# 提取基因符号列,并将其转换为字符向量
gene_vector <- as.character(gene_data[[1]]) ?# 将第一列基因符号列转换为字符向量
将基因符号转换为Entrez ID
# 使用bitr函数将基因符号转换为Entrez ID
# fromType = "SYMBOL" 表示输入的基因标识符是基因符号
# toType = "ENTREZID" 表示目标标识符是Entrez ID
# OrgDb = "org.Hs.eg.db" 指定使用人类基因注释数据库
entrez_ids <- bitr(gene_vector, fromType = "SYMBOL", toType = "ENTREZID", OrgDb = "org.Hs.eg.db")如下图所示为 entrez_ids 的转换结果,其中第一列为基因符号(symbol),第二列为对应的 ENTREZID 编号。
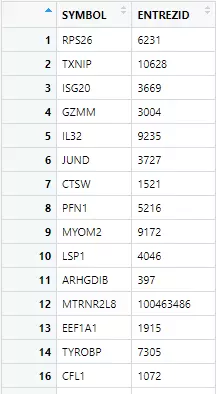
需要注意的是,在使用 bitr 函数进行基因 ID 转换时,部分基因可能无法匹配到相应的 ENTREZID,这种情况属于正常现象,主要由于数据库注释不完整或基因命名差异所致。
# 使用enrichGO函数进行基因本体(GO)富集分析
# gene = entrez_ids$ENTREZID:指定输入的基因列表,这里使用之前转换得到的Entrez ID
# OrgDb = "org.Hs.eg.db":指定使用人类基因注释数据库
# keyType = "ENTREZID":指定输入基因标识符的类型为Entrez ID
# ont = "ALL":指定分析所有三种GO本体(生物过程BP、细胞组分CC、分子功能MF)
# pAdjustMethod = "BH":指定使用Benjamini-Hochberg方法进行多重比较校正
# qvalueCutoff = 0.05:指定校正后的p值(q值)的阈值为0.05,只有q值小于该阈值的GO项才会被保留
# readable = TRUE:将输出结果中的ENTREZID转换为基因符号(SYMBOL)
go_enrichment <- enrichGO(gene = entrez_ids$ENTREZID,
OrgDb = "org.Hs.eg.db",
keyType = "ENTREZID",
ont = "ALL",
pAdjustMethod = "BH",
qvalueCutoff = 0.05,
readable = TRUE)
# 查看富集分析结果
# 使用print函数和summary方法打印GO富集分析的摘要结果
print(summary(go_enrichment))
# 将富集分析结果保存为CSV文件
# 使用write.csv函数将富集分析结果保存到指定路径的CSV文件中
write.csv(go_enrichment, file = "go_enrichment_results.csv", row.names = FALSE)
# 可视化 GO 富集分析结果
# 使用 dotplot 函数生成 GO 富集分析的点图
p <- dotplot(go_enrichment) # 生成点图对象
# 生成按本体(ONTOLOGY)分类的点图,并使用 facet_grid 分面显示
dotplot(go_enrichment, split = "ONTOLOGY") + facet_grid(ONTOLOGY ~ ., scale = "free")
# 使用 barplot 函数生成 GO 富集分析的柱状图
barplot(go_enrichment)KEGG 富集分析
# 使用 enrichKEGG 函数进行 KEGG 通路富集分析
# gene = entrez_ids$ENTREZID:指定输入的基因列表(Entrez ID)
# organism = "hsa":指定分析的生物物种为人类(hsa)
# keyType = "kegg":指定输入基因标识符的类型为 KEGG ID
# pAdjustMethod = "BH":指定使用 Benjamini-Hochberg 方法进行多重比较校正
# qvalueCutoff = 0.05:指定校正后的 p 值(q 值)的阈值为 0.05
kegg_enrichment <- enrichKEGG(gene = entrez_ids$ENTREZID,
organism = "hsa",
keyType = "kegg",
pAdjustMethod = "BH",
qvalueCutoff = 0.05)
# 将输出结果中的ENTREZID转换为基因符号(SYMBOL)
kegg_enrichment=DOSE::setReadable(kegg_enrichment, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
# 查看 KEGG 富集分析结果
print(summary(kegg_enrichment))
# 将 KEGG 富集分析结果保存为 CSV 文件
write.csv(kegg_enrichment, file = "D:\\Users\\Desktop\\call-task_KEGG_enrichment\\kegg_enrichment_results.csv", row.names = FALSE)
# 可视化 KEGG 富集分析结果
p1 <- dotplot(kegg_enrichment)
# 使用 barplot 函数生成 KEGG 富集分析的柱状图
barplot(kegg_enrichment)
 京公网安备 11010802022788号
京公网安备 11010802022788号







